Cagrilintide Sequence Cagrilintide
Introduction: Why the right cagrilintide sequence details matter
If you’re working with cagrilintide—whether for peptide research planning, analytical method development, or formulation trials—one frustration keeps coming up: small sequence-related differences can derail workflows. In my hands-on work, I’ve seen teams spend days chasing “mysterious” shifts in retention time, MS fragment patterns, or fragmentation behavior before realizing the root cause was a misunderstanding around the cagrilintide sequence they were actually working with.
This guide explains what the cagrilintide sequence is, why it drives analytical results and downstream behavior, and how to validate your understanding in a practical, lab-ready way. You’ll also find an FAQ with common questions researchers ask when they’re trying to reduce risk and rework.
What is cagrilintide (and where the sequence fits in)
Cagrilintide is a synthetic peptide used in research contexts, where its biological activity and analytical signature are tightly connected to its amino-acid ordering. In peptide work, the “sequence” isn’t just a label—it’s the blueprint that determines:
- Primary structure: the exact order of amino acids.
- Ionization and charge state behavior in mass spectrometry.
- Proteolytic digest patterns (how enzymes cut it).
- Fragmentation logic in tandem MS (MS/MS).
- Stability and handling considerations related to residues and motifs.
In my experience, when teams do not treat the cagrilintide sequence as a critical specification, they end up “testing until it works,” which is expensive and slow. A sequence-first approach makes your experiments measurable and reproducible.
How the cagrilintide sequence influences analytics (what you can actually observe)
When you run cagrilintide through common peptide characterization workflows, the cagrilintide sequence directly affects what you’ll see. Here’s how the logic usually maps to real lab outputs:
1) LC retention time and peak shape
While LC retention time is influenced by many factors (column chemistry, mobile phase, temperature), peptide primary structure drives hydrophobicity distribution and possible intramolecular interactions. In practice, when I troubleshoot retention time “drift,” I check sequence consistency alongside method parameters. If the sequence differs, even slightly, the chromatographic behavior can change enough to complicate peak assignment and purity estimates.
2) Mass spectrometry mass accuracy and isotope pattern
The theoretical monoisotopic mass is derived from the exact cagrilintide sequence. When you compare observed mass to theoretical mass, you’re essentially validating whether the peptide identity matches the sequence you believe you have. Small sequence mismatches typically produce mass errors far beyond your instrument’s tolerance.
For MS/MS, the sequence informs expected fragment ions (b/y ions) and their relative plausibility during fragmentation. If your fragmentation map doesn’t support the sequence, you may be dealing with:
- Sequence mismatch (wrong peptide, wrong batch spec, or labeling error)
- Unexpected modifications (e.g., incomplete synthesis issues or processing-related changes)
- Contaminants with similar intact mass or co-eluting species
3) Enzymatic digestion maps (proteomics-style confirmation)
If you digest cagrilintide with a protease (or in a controlled analytical digestion workflow), the amino-acid ordering determines cleavage opportunities and resulting peptide fragments. In my hands-on troubleshooting, digestion mapping is often the fastest way to confirm identity when intact-mass ambiguity exists due to adducts, salt effects, or near-isobaric impurities.
Using sequence-aware QA: a practical validation workflow
Instead of treating sequence verification as paperwork, I recommend building a sequence-aware QA loop into your workflow. Here’s a practical approach you can adapt to your lab constraints:
Step 1: Capture the sequence as a specification artifact
Create a single source of truth for the cagrilintide sequence you are planning and validating against. Store it alongside:
- Supplier/lot reference or internal lot identifier
- Planned analytical methods (LC and MS conditions)
- Expected theoretical mass and any reference fragment expectations
Step 2: Confirm intact identity before deep interpretation
Run intact identity checks early. If you’re doing MS, compare observed vs theoretical mass accuracy. If you see systematic deviations, it’s usually better to stop interpretation and resolve identity first—don’t “force-fit” results to your model.
Step 3: Use MS/MS or targeted digestion for sequence consistency
Once intact identity is plausible, confirm by either MS/MS fragment consistency or digestion mapping. My rule of thumb: if the sequence is central to your decision-making, you should have at least one orthogonal confirmation method that ties back to the cagrilintide sequence.
Step 4: Document method-to-sequence assumptions
Analytical methods often embed assumptions (ionization behavior, charge states, digestion efficiency). Documenting those assumptions upfront prevents confusion later—especially when multiple team members are working on different parts of the workflow.
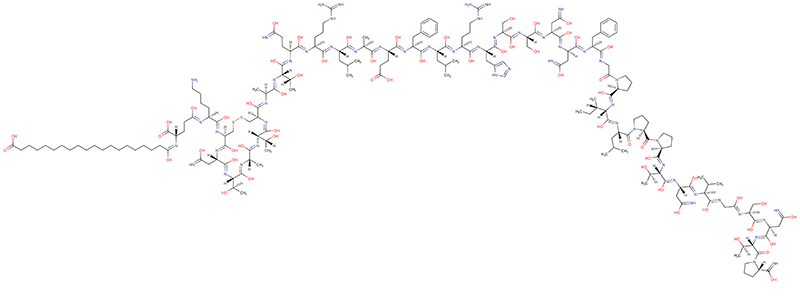
Common pitfalls when dealing with cagrilintide sequence information
Here are issues I’ve seen repeatedly in peptide projects—these are the reasons rework happens:
- Relying on a sequence copy without validation: If the sequence came from a document, label, or secondary source, confirm experimentally.
- Ignoring lot-to-lot variation context: Even when the intended sequence is stable, impurities or processing differences can affect purity and analytical interpretation.
- Comparing results using mismatched methods: LC and MS settings can change peak appearance and fragment patterns; you need method-consistent comparisons.
- Over-interpreting a single signal: One data point (e.g., one mass measurement) rarely settles identity when sequence is the key variable.
Being realistic about these pitfalls improves both speed and confidence. In research timelines, reducing uncertainty is often more valuable than squeezing extra sensitivity out of an instrument.
FAQ
What does “cagrilintide sequence” mean in peptide research?
It refers to the exact order of amino acids in the cagrilintide molecule. That primary structure determines theoretical mass, expected fragmentation/digestion patterns, and many analytical behaviors you’ll observe in LC-MS workflows.
How do I verify the cagrilintide sequence experimentally?
A practical approach is to confirm intact mass first, then validate sequence consistency using MS/MS fragment patterns or a targeted digestion/fragmentation map. Using an orthogonal method helps rule out coincidences caused by adducts or co-eluting impurities.
Why can two samples with similar handling show different analytical results?
Sequence-related differences (or unexpected modifications/impurities) can change ionization, fragmentation, and chromatographic behavior. Even if your method is “the same,” underlying identity or impurity composition can shift peaks and fragment distributions—so identity validation tied to the cagrilintide sequence matters.
Conclusion: Your next practical step
The cagrilintide sequence is more than a reference string—it’s a specification that governs identity confirmation, analytical interpretation, and the ability to reproduce results. If you anchor your workflow to sequence-aware validation (intact identity plus an orthogonal confirmation like MS/MS or digestion mapping), you reduce the common failure mode: spending time interpreting data that doesn’t match the peptide you think you have.
Next step: Build a single “sequence specification” record for your work (including expected theoretical mass), then run an early identity check (intact mass) followed by an orthogonal confirmation (MS/MS or digestion mapping) before making downstream decisions.
Discussion